NB. this content is reposted from an article I wrote for the NZSBMB "Southern Blot" newsletter.
I thought I’d talk about something else, something I find particularly interesting: Negative Controls4! I have worked with researchers who proudly talk about how many lanes of their gels are dedicated to their negative controls, and I’m sure all researchers spend a lot of time and effort on developing appropriate negative controls. Which is great. Yet, some researchers when they move from the wet-lab to sitting in front of a computer will forget about running negative controls on their bioinformatic experiments too. I did mention this issue in my talk in the context of our investigation into the interplay behind the speed and accuracy of bioinformatic tools3.
I thought I’d talk about something else, something I find particularly interesting: Negative Controls4! I have worked with researchers who proudly talk about how many lanes of their gels are dedicated to their negative controls, and I’m sure all researchers spend a lot of time and effort on developing appropriate negative controls. Which is great. Yet, some researchers when they move from the wet-lab to sitting in front of a computer will forget about running negative controls on their bioinformatic experiments too. I did mention this issue in my talk in the context of our investigation into the interplay behind the speed and accuracy of bioinformatic tools3.
Negative Controls are a backbone of research, and an important component of the definition of P-values that are frequently used to report the significance of findings 5 (the “null” distribution/hypothesis is of course a form of negative control).
But how many researchers run a negative control when they conduct a homology search (e.g. BLAST), or build a multiple sequence alignment (Clustal, MUSCLE, MAFFT), assemble a genome, map reads to a genome or predict the structure of a biomolecule of interest?
What is a reasonable negative control for these tasks? Randomised, reversed or circularly permuted sequences are popular instruments (this requires care too, as randomisation can be surprisingly difficult6). Biological controls may include approaches like trying to map E. coli reads onto a human genome as a control for a human RNA-seq experiment. There are a myriad number of possible controls, and it can sometimes take a lot of imagination and thought to find good ones.
These are very worthwhile, as the results of negative controls can be surprisingly informative. They can not only allow us to reject null hypotheses and compute P-values, but can help identify false positives, contamination, biases or even identify unanticipated signals in our data.
Some bioinformatic tools will try to give an idea of the significance of different results. E-values are a popular measure of significance, these are the expected number of matches of a score as good or better, for a known query and database size. Most variants of BLAST, for example, will report an E-value for each match7. These E-values are estimated using the following formula:
E = Κmn e-λx
Where ‘x’ is the alignment bitscore, ‘m’ and ‘n’ are the size of your search database, and ‘Κ’ and ‘λ’ are generic fitting parameters to model the distribution of scores expected between non-homologous sequences. BLAST attains much of it’s speed by using pre-computed values of ‘Κ’ and ‘λ’ for typical selections of search parameters.
As widely used as these E-values are, they can be misused and misleading in a number of ways. Two of which I’ll discuss below.
How many times do we conduct multiple searches in a single experiment? Now, how often do we correct E-values for the multiple searches that we have run? I’ve got to admit, I have never done this. Many of us know about the statistical sin of uncorrected multiple-testing8, where multiple comparisons may be made but the P-values are computed assuming a single comparison. For example, we may wish to test if there is a correlation between the expression of a particular gene and the expression of other genes. If we use a P-value threshold of 0.05, then 5% of the time we expect to falsely reject the null (i.e. report a significant finding), if we run 10,000 tests assuming each time that a single test is being run (e.g. ‘cor.test()’ in R), then we can expect to report 500 falsely significant results. Since there is a straightforward correspondence between E-values and P-values7 (P = 1-e-E), then we really should be correcting our E-values as well as our P-values.
A common practice is to compare the entire proteome of one organism against that of another organism (e.g. E. coli and Salmonella typhi) or it is more common these days to compare hundreds or thousands of bacterial proteomes at a time. If we’re using E-values to evaluate the significance of results, then a correction of the E- or P-values should be made. Commonly used methods may include Bonferroni or the Benjamini–Hochberg, each of which may provide a better estimate of the true significance of your results. Alternatively, another strategy that may be more informative, yet uses a lot more computational resources, is to run the same queries with randomized sequences and use these results to evaluate the significance of one’s results. The distribution of (bit) scores resulting from these searches will help establish the significance of the “real” matches.
Another common mistake than can compound errors when working with E-values is to use ‘-log(E)’ values as a summary statistic. This value is used by the popular orthoMCL tool for finding homologous clusters resulting from all-vs-all comparisons of proteomes9,10. Computationally, the all-vs-all comparison is an extremely costly procedure. It is therefore surprising to me that ‘-log(E)’ is used for assessing the relationships between proteins. If you take the ‘-log’ of the above definition of ‘E’ and use a little algebra, you will hopefully see that that ‘-log(E)’ is a conversion back to a bitscore with some other terms that may accentuate, rather than correct problems due to database and query size (i.e. the larger the database or query, the larger a chance bitscore is likely to be). In fact, it was recently shown empirically that ‘-log(E)’ values perform worse than unadulterated bitscores for sequence clustering11.
In order to illustrate how it is possible to find matches between randomised sequences and large sequence databases I have selected sigma factors from E. coli and used randomised versions of these as queries for the NCBI BLAST and EBI HMMER webservices. Each method reported matches to my queries, the E-values associated with these are not meaningful (due to multiple testing, as described above), therefore I have only reported the bit scores (Figure 1).
BLASTP:
38.0 bits
Query 301 GEVETSIPRVDVQENNSNNQLRQLLALSLLIELPLDNVQQLGSMPPISYSAFDLGEMNLT
G E ++R+D ++ + + L+++ + ++ +++++G++P + ++ L + + Sbjct 22 GRAEVMLARLDAGTLDAAQLRKGLVSIREMTRDAIVQLERMGPLPRATEAP--LPAVLVV
PHMMER:
18.5 bits
QUERY yeap.rtvhhq.dqtksdlealesnhlesqtkmsspivgvllldgrtttsvdlategaietkdvqq
MATCH y p r +h q d + d e e+ hl++ + +++v+ +d+rt +v+l+ + +ietkd q
PP 55553678877255589**********8765..56789*************************875
TARGET YATPeRQLHLQtDTDRCDSETSENQHLQNIE--NQAMINVVAIDNRTPQNVNLSKDHSIETKDQQS
JACKHMMER:
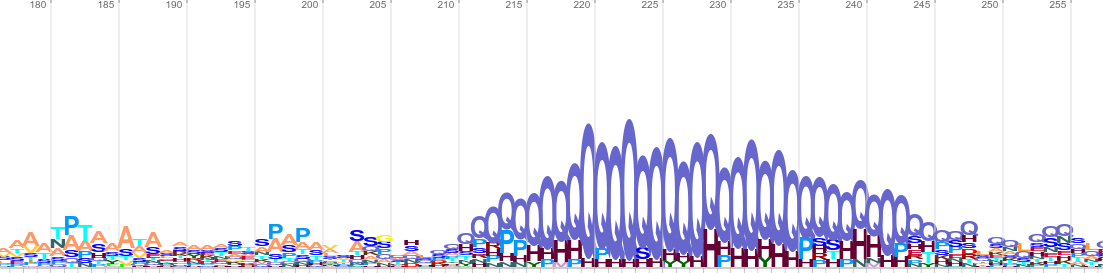 |
Figure 1: Alignments resulting from homology searches with randomised E. coli sigma 54 (RpoN) sequences using the NCBI webservice for BLASTP and the “nr” database (April 2017), the EBI webservice for PHMMER (v3.1b2) and the “reference proteome” database, and a fragment of a sequence logo generated after four iterations of the EBI’s JACKHMMER (v3.1b2) webservice.
Should I only pick on BLAST and other homology search tools? BLAST is one of the work-horses of bioinformatics, a popular tool for many pipelines, and it’s original description is one of the world’s most cited papers. Unlike homology searches, multiple sequence alignment is almost entirely free of any sort of significance assessment. Therefore we have some reports of terrifying statistics regarding multiple sequence alignment 12. Bradley et al. conclude from a randomized sequence alignment test that “for most alignment programs, aligned sequences are not necessarily homologous”. In fact, popular alignment tools like ClustalW will align 95 to 97% of randomised (non-homologous) sequences that are given as input. This is worrying as multiple sequence alignment is the first step before many phylogenetic, genomic, evolutionary and structural analyses. Most of the popular tools for this assume that the input sequences are in fact homologous, this can result in superficially robust results that are indistinguishable from randomly generated results (Figure 2).
 | 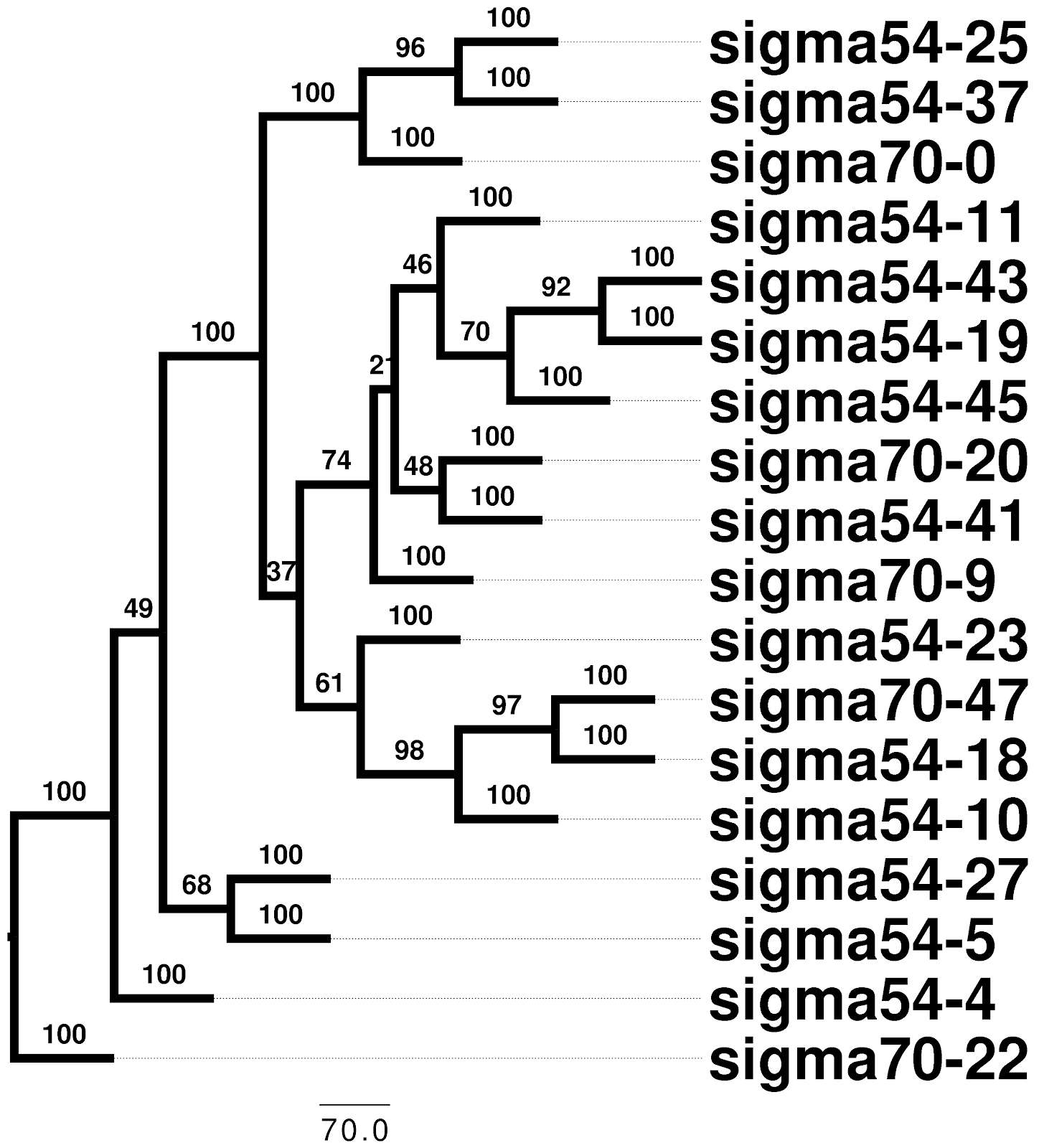 |
Figure 2: Randomised sigma factor sequences from E. coli were clustered using MAFFT (v7.271) alignments and distances from “protdist” (PHYLIP v3.696). A “clade” of sequences was extracted (left) and a phylogenetic tree for these was inferred using RaxML with the “PROTGAMMAWAG” model, 100 bootstrap replicates were used to establish the robustness of tree edges, a consensus tree was built using “consense” from PHYLIP (right).
We can run similar experiments and show similar issues with falsely significant findings with all the sequence assembly, read mapping, expression analysis, metagenomics, proteomics and bioinformatic tools we use to analyse bioinformatic data.
Closing statements
As our ability to generate larger datasets, our databases, and our computational resources continue to grow exponentially, it is a good idea to question the likelihood of results, using genuine and realistic negative controls. This is because, as sequence space grows, the number of false positives increase, meaning that calibrations that worked decades ago are increasingly irrelevant.
Some of our other assumptions regarding genomics are also worth revisiting. How biologically meaningful are the data from the sequences we assemble, the comparisons we make, the RNA-seq reads, the protein-nucleotide interactions and the myriad other datasets we collect on a daily basis? Sean Eddy proposed a fascinating experiment, the “Random Genome Project” 13. This project acknowledges that cells are noisy environments, packed full of biomolecules that jostle together, and random, non-functional interactions and events constantly occur. In this context a useful control to have, is a synthetic “random” chromosome that has been inserted into your organism of interest. Then all outputs, RNA, protein and interactions that map to your random chromosome serve the invaluable, yet missing null distribution for genomics projects. I’ve not seen a project that has taken on Sean’s challenge yet, but I’m very much looking forward to seeing the results.
Note added:
This paper comes close to the "random genome project" idea:
Neme et al. (2017) Random sequences are an abundant source of bioactive RNAs or peptides.
Acknowledgements
Thanks to Bethany Jose and Nicole Wheeler for proofreading the draft and lively research discussions. A huge thanks to the clever, fun and inspiring community of students and collaborators that I get to work with.
References
1. Gardner, P. Random RNA interactions control protein expression in prokaryotes. (2016). Available at: https://www.slideshare.net/ppgardne/random-rna-interactions-control-protein-expression-in-prokaryotes. (Accessed: 2nd April 2017)
3. Gardner, P. P. et al. A meta-analysis of bioinformatics software benchmarks reveals that publication-bias unduly influences software accuracy. bioRxiv 092205 (2017). doi:10.1101/092205
4. Wikipedia contributors. Scientific control. Wikipedia, The Free Encyclopedia (2017). Available at: https://en.wikipedia.org/w/index.php?title=Scientific_control&oldid=768804853. (Accessed: 1st April 2017)
5. Wikipedia contributors. p-value. Wikipedia, The Free Encyclopedia (2017). Available at: https://en.wikipedia.org/w/index.php?title=P-value&oldid=771502462. (Accessed: 1st April 2017)
6. Wikipedia contributors. Fisher–Yates shuffle. Wikipedia, The Free Encyclopedia (2017). Available at: https://en.wikipedia.org/w/index.php?title=Fisher%E2%80%93Yates_shuffle&oldid=771535751. (Accessed: 1st April 2017)
7. The Statistics of Sequence Similarity Scores. Available at: https://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html. (Accessed: 1st April 2017)
8. Wikipedia contributors. Multiple comparisons problem. Wikipedia, The Free Encyclopedia (2017). Available at: https://en.wikipedia.org/w/index.php?title=Multiple_comparisons_problem&oldid=770714321. (Accessed: 1st April 2017)
13. Eddy, S. R. The ENCODE project: missteps overshadowing a success. Curr. Biol. 23, R259–61 (2013).
No comments:
Post a Comment